Population Genomics Modeling
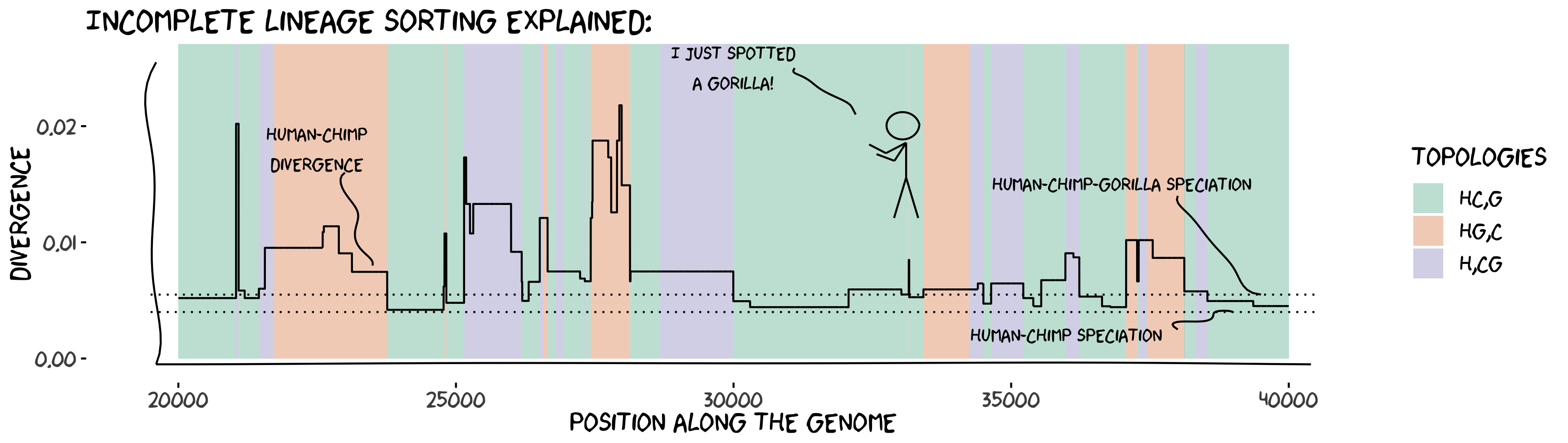
Modeling the evolution of genomes in populations enables predicting the patterns of genetic diversity along the genome and between species. Combined with statistical inference methods, population genetic modeling confronts the predicted patterns with those observed in datasets of genomes in natural populations, permiting the inference of key parameters (demographic history, recombination landscapes, selection map, etc.), as well as testing of hypotheses regarding evolutionary processes by model comparison.
The theoretical framework that makes these analyses possible is the coalescent theory, and, as far as sexually reproducing species are concerned, the so-called coalescent with recombination. In the presence of recombination, the history of a sample can be represented as a complex structure called an <ancestral recombination graph(ARG). The ARG is not directly measurable from the data, as many different ARGs can generate the same sequences. Inference methods, therefore, need to integrate over all possible ARGs - a computationally demanding procedure that prohibits the analysis of large sequences, a fortiori complete genome data.
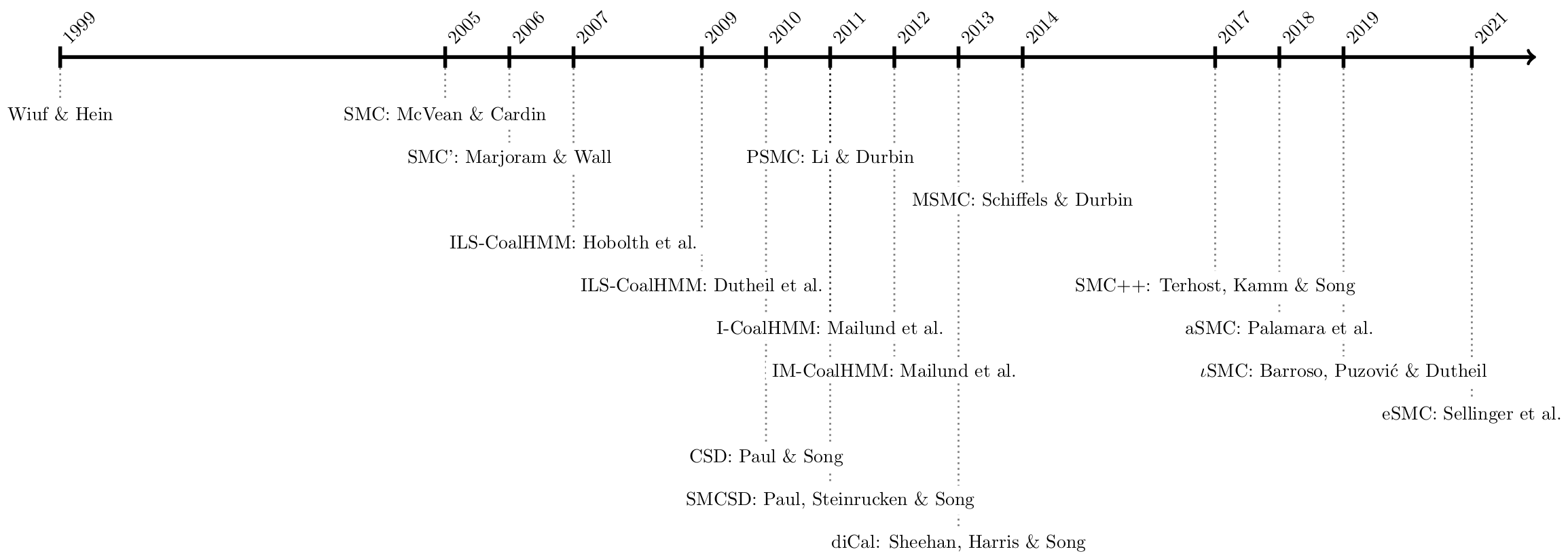
The sequentially Markov coalescent (SMC) is a model that approximates the coalescent with recombination. The model as been developed and used over le last 20 years (Figure 1). By ignoring certain types of recombination events, the SMC is a markovian process along the genome: the genealogy at one position can be directly modeled from the genealogy at the previous position. This property allows using hidden Markov models to efficiently integrate over the ARG and infer model parameters over complete genome datasets. This approach has been termed coalescent hidden Markov model (CoalHMM).
A large set of SMC-based models has been developped, including increasingly complex demographic scenarios.
These models, however, assume that the process is homogeneous along the analysed sequences.
As the genealogical process depends, in part, on the recombination rate, this assumption is at odds with the large body of evidence for heterogeneous recombination landscapes.
To address this issue, we have developped and extension of the SMC that allows some parameters to vary along the genome
.
The integrative sequentially Markov coalescent, iSMC
The HMM implementation of the SMC process for two genomes (Pairwise Sequentially Markov Coalescent, PSMC) depends on a transition matrix that is homogeneous along the analyzed sequences.
The underlying transition probabilities depend on the demographic model (variation of population sizes in time) and a genome average recombination rate.
We introduced a new modeling framework allowing any parameter to vary along the genome
.
To do so, we consider that heterogeneous parameters follow an a priori distribution (which can have free parameters), and that the parameter values are auto-correlated along the genome, in a Markovian manner.
As a result, the SMC become a Markov-modulate Markov process, which can be rewritten as a Markov process where the states are combinations of divergence classes (as in the PSMC) and heterogeneous parameter values.
This methodology can then be used to infer site-specific parameter values, and was first applied to the inferrence of recombination rates
by Dr Gustavo Barroso during his PhD work, providing the first method to provide recombination map with a single, unphased, diploid genome.
This allowed us to infer the recombination map of several ancient hominids.
We further extended the model to account for both variable recombination and mutation rates
. This modeling framework allowed us to jointly estimate the contribution of mutation rate variation and processes affecting the ARG to observed genetic variation
. Using extensive simulations and a population genomic dataset of the fruit fly Drosophila melanogaster, we showed that mutation rate variation is the main driver of genetic variation along the genome. Probably because the mutational landscape of a genome is a very difficult data to obtain, very few studies aimed at assessing its impact and focused instead on understanding how much selection vs. genetic drift, or how much background selection vs. genetic draft, explained the diversity not accounted by mutation rate variation. iSMC offers a new framework permiting to add mutation rate variation to the picture.
References
- Dutheil JY. Towards more realistic models of genomes in populations: The Markov-modulated sequentially Markov coalescent, in Probabilistic Structures in Evolution. European Mathematical Society Publishing House, 383408.
- V Barroso G, Puzović N, Dutheil JY. Inference of recombination maps from a single pair of genomes and its application to ancient samples, . PLoS Genetics 15(11): e1008449.
- V Barroso G and Dutheil JY. The landscape of nucleotide diversity in Drosophila melanogaster is shaped by mutation rate variation, . Peer Community Journal 3: e40.